Panacea Journal of Medical Sciences
Panacea Journal of Medical Sciences (PJMS) open access, peer-reviewed triannually journal publishing since 2011 and is published under auspices of the “NKP Salve Institute of Medical Sciences and Research Centre”. With the aim of faster and better dissemination of knowledge, we will be publishing the article ‘Ahead of Print’ immediately on acceptance. In addition, the journal would allow free access (Open Access) to its contents, which is likely to attract more readers and citations to articles published in PJMS.Manuscripts must be prepared in accordance with “Uniform requiremen...

A case series of heriditary spastic paraplegia
Abstract
Hereditary spastic paraplegia is a spectrum of disorders with progressive spasticity of lower limbs with clinical and genetic heterogeneity. Harding classified HSP into pure and complicated forms. Currently classification is largely based on genetics. Around 87 genetic loci were identified till date. We have studied 44 patients presented with spastic ataxia with or without additional neurologic or non-neurological features from February 2021 to February 2024. Out of 44 spastic ataxia cases studied over period of 3 years, 17 patients were positive for SPG genes with 21 variants detected by NGS method. New phenotypic representation of SPG 28 as it can present with spastic paraparesis and distal upper limb amyotrophy, i.e., Silver Syndrome. We found SPG 11 to be the most common inherited cause of hereditary spastic paraplegia in our study. SPG 7 is next common HSP type found. In conclusion, SPG 11 should be tested in cases presenting with spastic paraparesis before second decade, with thin corpus callosum, ear of lynx on MRI along with other neurological features like cognitive decline. With gene specific therapy on the horizon diagnosing HSP cases based on NGS should be considered.
Introduction
Hereditary spastic paraplegia is a spectrum of disorders with progressive spasticity of lower limbs being a common finding, with clinical and genetic heterogeneity. Prevalence of HSP was found to be 3.6 per 1, 00,000 population all over the world.[1] Harding classified HSP in to pure and complicated forms based on the presence of UMN paraparesis alone and in combination with other features respectively. Now classification is largely based on genetics. Around 87 genetic loci were identified till date.
Materials and Methods
We have studied 44 patients presented with spastic ataxia with or without additional neurologic or non-neurological features from February 2021 to February 2024. Deep phenotyping with a detailed pedigree charting was done. MRI brain and spine study was done in all patients .NCS, EEG studies were done wherever appropriate. Genetic studies (WES by NGS, PCR for repeat disorders) were done based on phenotype and investigations .Out of 44 spastic ataxia cases studied over period of 3 years, 17 patients were positive for SPG genes with 21 variants detected by NGS method. Patient with mutation in SPG loci were studied further to understand clinical and genetic spectrum.
Results
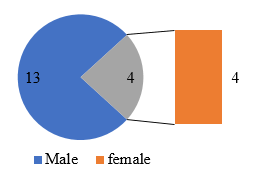
Out of 44 spastic ataxia cases, a total of 17 genetically confirmed cases of hereditary spastic paraplegia were studied. Out of 17, 13 were male (76.47%) and 4 (23.53 %) were female patients ([Figure 1]). Most common age of onset was found to be 2nd and 3rd decade (47 %). Late onset hsp was found in 3 cases in our study. One Patient presented at 5th decade, with onset of symptoms at 48 years of age. Two patients presented in 4th decade with 33- and 35-years age of onset.
Third degree Consanguinity is found in 5(29.41%) cases in our study. ([Figure 2], [Figure 3]). Positive Family history was found in 6 patients (35.29%). Family history is autosomal recessive like in all these patients .Among the patients with positive family history, five families had patient and their siblings affected. Only in one family, father and son were affected.
In our cohort, Pure HSP is seen in 3 (17.64%) cases, complicated HSP in 14 (82.35 %) cases. Out of 14 complicated HSP cases, 6 had cognitive decline (35%) along with spasticity. Likewise, dysarthria in 3 (17.6%), Extrapyramidal involvement in 3(17.64%) (2 had dystonia, 1 had parkinsonian features), ataxia in 1 (6.67%) patient was found.
On imaging studies, specific pattern corresponding with HSP was found in 8 patients. MRI brain and spinal cord done in all patients, out of which, thin corpus callosum is seen in 4 patients, Ear of lynx sign is seen in 2 patients; all of them were found to be SPG 11. Cervicodorsal cord atrophy seen in SPG 10 and SPG 30. Bilateral periventricular hyperintensities were seen in 2 cases. Cerebellar atrophy seen in 2 cases, that is SPG 15 and 35. MRI was normal in 5 cases. Electrophysiology studies showed neuropathy in 2 patients and amyotrophy (motor neuron disease) in 1 patient.
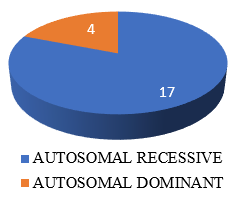
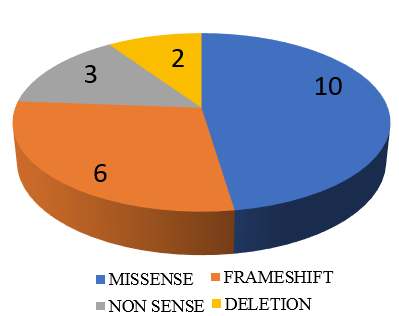
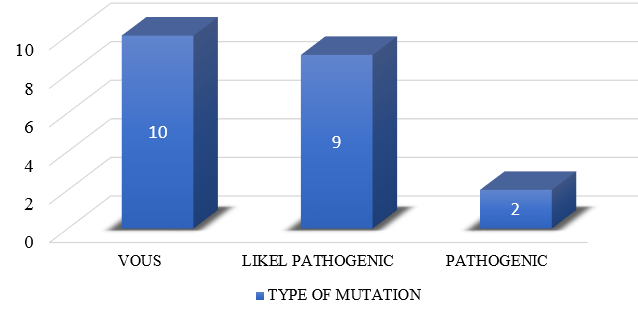
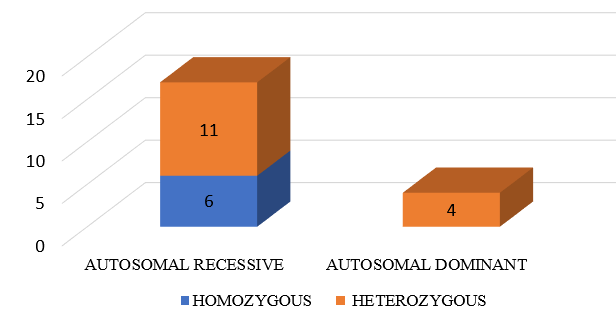
Out of 21 mutations detected in SPG loci, 9 were Likely pathogenic cases (percentage), 2 patients had Pathogenic and 10 patients had variant of unknown significance mutations. ([Figure 5], [Figure 6]). Out of these 21 SPG loci mutations detected, 10 missense mutations, 6 frame shift mutations (2 AMINO ACID SUBSTITUTIONS, 2 DELETIONS ,1 LOSS OF FUNCTION), 3 nonsense mutations and 2 deletions were seen. We have seen the inheritance pattern to be compound heterozygous autosomal recessive in 11, homozygous autosomal recessive in 6, autosomal dominant in 4. Out of 14 complicated hsp cases, recessive pattern of inheritance is seen in 12 and dominant in 2. Of the 3 pure hsp cases in our study, 2 have autosomal recessive pattern of inheritance and 1 has dominant inheritance.
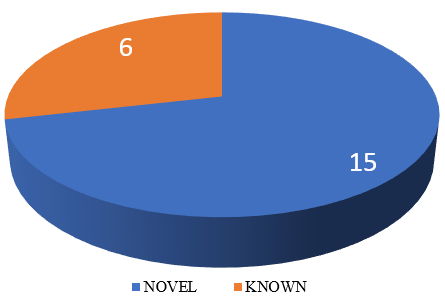
Out of 21 SPG loci detected, we found c.3623C>T (p. Pro1208Leu) mutations common in 2 different unrelated patients i.e., in 22-year-old female patient from Uttar Pradesh and an 18-year-old male patient from Maharashtra. Another Homozygous autosomal recessive nonsense mutation at chr15:44865850G>A c.6100C>T p. Arg2034Ter loci is found in 2 unrelated patients. Autosomal dominant mutation was found in SPG17/ SPG 73, SPG 42, SPG 30, SPG 10/ SPG 7. 15 novel mutations were found in our study ([Figure 4]).
|
S.No |
Age of onset |
Age of presentation |
Sex |
Description |
Consanguinity |
Other features |
Family hsitory |
MRI |
NCS |
|
1 |
25 Y |
29 Y |
F |
UMN Type of weakness of both L/L and bladder/Bowel dysfucntion |
Non consanguinous |
NIL |
No |
Normal |
Normal |
|
2 |
3 Y |
10 Y |
M |
Progressive quadriparesis |
Non consanguinous |
Delayed milestones |
No |
Dorsal cord atrophy |
Peripheral neuropathy |
|
3 |
25 Y |
25 Y |
M |
B/L L/L UMN Weakness |
Non consanguinous |
Nil |
No |
Periventricular hyperintensities |
Normal |
|
4 |
17 Y |
22 Y |
F |
UMN B/L L/L Weakness |
Non consanguinous |
Cognitive involvement seizures |
No |
Thin corpus callsoum |
Normal |
|
5 |
48 Y |
50 Y |
F |
B/L L/L UMN Type of weakness |
Non consanguinous |
Backward falls, Weight loss |
No |
Dorsal cord atrophy |
Peripheral neuropathy |
|
6 |
16 Y |
19 Y |
M |
B/L L/L UMN Weakness |
Non consanguinous |
Cognitive decline |
Brother affected |
Ear of lynx |
Normal |
|
7 |
14 Y |
19 Y |
M |
B/L UMN Weakness and imbalance while walking |
Consanguinous |
Cognitive decline, Parkinsonism, pseudo bulbar |
2 brothers affected |
Thin corpus callsoum |
Normal |
|
8 |
33 Y |
37 Y |
M |
B/L L/L UMN Weakness with urinary urgency, Dysarthria |
Non consanguinous |
Urinary urgency, Dysarthria |
No |
Dorsal cord atrophy |
Normal |
|
9 |
21 Y |
25 Y |
M |
Dystonic myoclonic tremors, spastcity of bpoth l/l , ataxia |
Consanguinous |
Dystonic myoclonic tremors |
No |
Cerebellar atrophy |
Normal |
|
10 |
25 Y |
28 Y |
M |
B/L U/L Distal UMN Weakness since 3 years, L/L Weakness since 1 year |
Non consanguinous |
U/L Distal amyotrophy |
No |
Normal |
Normal |
|
11 |
5 Y |
18 Y |
F |
B/L L/L UMN Weakness |
Non consanguinous |
Cognitve decline |
Elder sister effected |
Normal |
Normal |
|
12 |
35 y |
40 Y |
M |
LBA , B/L UMN Weakness |
Non consanguinous |
Nil |
Son affected |
Normal |
Normal |
|
13 |
2 y |
20 Y |
M |
B/L L/L UMN Weakness |
Non consanguinous |
Poor scholastic performace |
Eleder broither effected |
Normal |
Normal |
|
14 |
8 Y |
18 Y |
M |
B/L L/L UMN Weakness |
Consanguinous |
Cognitive decline |
No |
Thin corpus callsoum |
Normal |
|
15 |
13 Y |
23 Y |
M |
B/L L/L UMN Weakness, Urinary retention |
Non consanguinous |
Dysarthria, Urinary retention |
No |
Ear of lynx |
Normal |
|
16 |
5 Y |
7 Y |
F |
B/L LIMB Ataxia, B/L L/L UMN |
Consanguinous |
Dystonia in L/L, Flat foot |
No |
B/l Periventricular hyperintensities |
Normal |
|
17 |
2 Y |
6 Y |
MALE |
B/l l/l Stiffness and imbalance while walking |
Consanguinous |
Ataxia,flat foot |
younger brother affected |
Cerebellar atrophy |
Normal |
|
S.No |
GENE |
EXON |
Variant |
Type of mutation |
Pathogenic/ VOUS |
Novel/ Known |
Inheritance pattern |
HSP |
|
1 |
CPT1C , BSCL2 |
EXON 18, EXON 7 |
C.1994C>T (p.Ser66gLeu), C.754T>C (p.Phe252Leu ) |
Missesnse, missense |
VOUS, VOUS |
Known, novel |
Heterozygous autosomal dominant, heterozygous autosomal dominant |
SPG73, SPG17 |
|
2 |
KIF1A |
EXON 45 |
C.4781C>T p.Ser1594Leu |
Missense |
VOUS |
Known |
Heterozygous autosomal dominant/ recessive |
SPG 30/ Hereditary sensory neuropathy type IIC |
|
3 |
CACNA1G , PNPLA6 |
EXON 13, EXON 25 |
C.2860G>A (p.Val954Met), c.2962G>A ( p.Ala988Thr) |
Missense, missense |
VOUS, VOUS |
Novel, novel |
Heterozygous autosomal dominant, heterozygous autosomal recessive |
SCA42, SPG39 |
|
4 |
SPG11 |
EXON 21 |
C.3623C>T (p.Pro1208Leu) |
Missense |
VOUS |
Novel |
Heterozygous autosomal recessive |
SPG11 |
|
5 |
KIF5A , SPG 7 |
Intron 26 , EXON 3 |
C.2993-1G>A , c.322C>G (p.Gln108Glu) |
Frameshift, missense |
Likely pathogenic, VOUS |
Novel, novel |
Heterozygous autosomal dominant, heterozygous autososmal dominant |
HSP10 , HSP7 |
|
6 |
SPG11 |
EXON 31 |
C.5939delt(p.Leu1980Argfs*78) |
Frameshift |
Likely pathogenic |
Novel |
Heterozygous autosomal recessive |
SPG11 |
|
7 |
SPG11 , SPG11 |
EXON 32 |
C.6658_6659delat p.Met2220fs*27, c.6100C>T p.Arg2034* |
Frameshift, nonsense |
Likely pathogenic, Pathogenic |
Novel, novel |
Heterozygous autosomal recessive, heterozygous autosomal recessive |
SPG11, SPG11 |
|
8 |
SPG7 |
EXON 6 |
C.778A>T p.Met260Leu |
Missense |
VOUS |
Novel |
Heterozygous autosomal recessive/autosomal dominant |
SPG7 |
|
9 |
RAPGEF2, ZFYVE26, ZFYVE26 |
EXON 7 |
C.4411C>T p.Pro1471Ser, c.6744_6746delgaa p.Lys2248DEL, c.1093delc p.Leu365Serfs*16 |
Missense, inframe deletion , frameshift |
VOUS, VOUS, Likely pathogenic |
Known, novel, novel |
Heterozygous autosomal dominant, heterozygous autosomal recessive, heterozygous autosomal recessive |
Familial adult myoclonus 7 , SPG 15 , SPG15 |
|
10 |
HSPB3, DDHD1 |
EXON 1 |
C.224T>A p.Leu75Gln, c.424delg p.Glu142fs*18 |
Missense, frameshift |
VOUS, Likely pathogenic |
Known, novel |
Heterozygous autosomal dominant, homozygous autosomal recessive |
HMN2C, SPG 28 |
|
11 |
SPG11 |
EXON 32 |
Chr15:44865850G>A c.6100C>T p.Arg2034Ter |
Nonsense |
Pathogenic |
Known |
Homozygous autosomal recessive |
SPG11 |
|
12 |
SPG7 |
EXON 2 |
Chr16:89576947T>A c.233T>A p Leu78Ter |
Nonsense |
Likely pathogenic |
Known |
Homozygous autosomal recessive |
SPG7 |
|
13 |
GBA2 |
EXON 9 |
C.1460T>C p.Leu487Pro |
Missense |
Likely pathogenic |
Novel |
Homozygous autosomal recessive |
SPG 46 |
|
14 |
SPG11 |
EXON 21 |
C.3623C>T p.Pro1208Leu |
Missense |
VOUS |
Known |
Homozygous autosomal recessive |
SPG11 |
|
15 |
SDHA,SPG11, SCN4A |
EXON 21-24 |
Chr5:251555G>A c.1766G>A p.Arg589Gl , chr15:c.3520+1_3521-1 )_(4161+1_4162-1)del, chr17:62022916C>T c.3524G>Ap.Ser1175aSN |
Missense, deletion, missense |
Likely pathogenic, Likely pathogenic, VOUS |
Known, novel, known |
Heterozygous dominant/Recessive, heterozygous autosomal recessive, heterozygous dominant/ Recessive |
SDHA, SPG11, CMS/hyperpp, hypopp |
|
16 |
SPG11, FLVCR1 |
EXON 15 |
C.2626A>A/G p.Lys876Glu, c.49C>C/G p.Pro17Ala |
Missense |
VOUS, VOUS |
Known, known |
Heterozygous missense autosomal recessive, heterozygous missense autosomal recessive |
SPG11, Posterior column Ataxia withrp |
|
17 |
FA2H /SPG 35 |
EXON 6 |
C.815duptp.Val275fs*38 |
Frameshift |
Likely pathogenic |
Novel |
Homozygous autosomal recessive |
FA2H / SPG35 |
|
Clinical features |
Our study |
SHI et al., Study [2] |
|
Cognitive decline |
6 (35.29%) |
6 (22.2%) |
|
Extrapyramidal |
3 (17.64%) |
3(11.1%) |
|
Dysarthria |
2 (11.7%) |
9(33.3%) |
|
Peripheral neuropathy |
2 (11.7%) |
3(11.1%) |
|
Ataxia |
1 (5.8%) |
3(11.1%) |
|
Seizure |
1 (5.8%) |
2(7.4%) |
|
S.No. of patients |
MRI Findings |
|
1 |
Normal |
|
2 |
Dorsal cord atrophy |
|
3 |
Periventricular hyperintensities |
|
4 |
Thin Corpus Callosum |
|
5 |
Dorsal cord atrophy |
|
6 |
EAR OF LYNX |
|
7 |
Thin Corpus Callosum |
|
8 |
Dorsal cord atrophy |
|
9 |
Cerebellar atrophy |
|
10 |
Normal |
|
11 |
Normal |
|
12 |
Normal |
|
13 |
Normal |
|
14 |
Thin Corpus Callosum |
|
15 |
EAR OF LYNX, Thin Corpus Callosum |
|
16 |
B/L Periventricular hyperintensities |
|
17 |
Cerebellar atrophy |
|
|
Our study |
Narendiran et al[3] (2022) |
Kamate et al.,[4] (2019) |
Stevanin et al. [5] (2008) |
Kara et al. [6](2016) |
Ishiura et al. [7] (2014) |
Luo et al. [8] (2014) |
Elert-Dobkowska et al.[2] (2019) |
|
Ethinicity |
India |
India |
India |
France |
UK |
Japan |
China |
Poland |
|
Cohort |
44 (17) |
57(25) |
11 |
76 |
97 (48) |
129(46) |
201 |
30 |
|
Age |
2-50 Y |
3-22 Y |
21.7 months. |
2 to 27 |
16 (3-39) |
10-50 |
SPG4 - 11-41 Non SPG4 5-34 |
NA |
|
Gender M:F |
13: 4 |
40:17 |
6:5 |
18:20 |
|
75:54 |
134:67 |
NA |
|
Pure or complicated |
3/14 |
15/42 |
4/7 |
0/38 |
|
0/97 |
130/71 |
16/14 |
|
Inheritance |
AR |
AR |
AR 10/ AD 1 |
AR (65%) |
AR |
AD |
AD |
AR |
|
GENE |
SPG11 |
SPG11 |
DDHD2 |
SPG11 |
|
|
|
|
Discussion
In our study, we found HSP more commonly in 2nd and 3rd decade (7-50 Y) with male predilection ([Table 1], [Table 2]) .Age of onset and sex predilection was in concordance with another Indian study i.e., Narendiran S. et al. [3] (3-22 Y). Similar age of presentation is found in Kara et al. [6] (3-39). Stevanin G et al. [5] study found to have more female preponderance with 2nd and 3rd decade presentation (2 to 27). Positive Family history is found in 6 patients. Most of the patients with family history had their siblings affected in same generation (5 patients). While in only one family, Father and son were affected with similar phenotype. Consanguineous marriage increases the frequency of AR HSPs in the community. Study by Arun Meyyazhagan et al. [9] found that European and north American population had predominance of autosomal dominant HSP whereas middle eastern and American amish groups had predominant autosomal recessive inherited hsp, attributed to the higher prevalence of consanguinity in these populations.
PURE HSP in our study is seen in 3 (17.64%), COMPLICATED HSP in 14 (82.35 %) cases. This is Corroborated by an Indian study by Narendiran S. et al., [3] where they found 17 complicated HSP and 7 pure HSP cases out of 24 genetically confirmed hsp cases. Unlike a chinese study by Cao, Y et al., [10] of a total of 270 patients, they found 67% of patients with pure HSP, and 33% with complicated HSP. European study by Elert-Dobkowska E et al., [2] in Poland, found pure HSP in 16 and complicated HSP in 14 out of 30 cases studied. In our study, predominance of complicated HSP is probably due to greater number of autosomal recessive HSP compared to autosomal dominant HSP.
We found cognitive decline to be the next most common clinical feature after spasticity of limbs, seen in 6 patients, extra pyramidal involvement in 3, dysarthria in 3, ataxia in 1 patient,Peripheral neuropathy is seen in 2 and upper limb distal amyotrophy in one patient. In Shi Y et al study., [11] dysarthria and cerebellar ataxia were detected in 9(33.3%) and 3(11.1%) of patients with HSPs, respectively. Comparison of clinical features with Shi Y et al. study [11] is shown in [Table 3]. Harding found urinary symptoms, sensory followed by ataxia to be the most common in the descending order of frequency. A 28-year-old male, born of non-consanguineous parents presented with 3 years progressive spastic paraparesis and upper limb distal amyotrophy. WES revealed DDHD 1 mutation, in SPG 28 loci. HSP with amyotrophy is not described in SPG 28 till date. All over world, only 3 cases of spg 28 reported to the best of our knowledge. Tesson et al. (2012) [12] reported 3 patients from 2 unrelated families with SPG28. Two Turkish brothers, born of consanguineous parents, showed progressive spastic gait with onset around adolescence. An unrelated 62-year-old French woman had spastic paraplegia since infancy and had an axonal neuropathy. None of them were reported to have amyotrophy. Spastic paraparesis with distal amyotrophy is called as silver syndrome. Phenotype of silver syndrome was described in SPG 17, SPG 38 and in few SPG 4 cases. We therefore describe a new phenotypic representation of SPG 28 i.e., spastic paraparesis with distal amyotrophy, called as silver syndrome.
Another 10-year-old male presented with progressive spastic quadriparesis along with peripheral neuropathy. On WES by NGS method, revealed mutation at KIF1A gene at c.4781C>T p.Ser1594Leu loci which can cause both SPG 30 and HEREDITARY SENSORY NEUROPATHY TYPE IIC. Mutation at one locus, with overlapping clinical manifestation is seen in this case.
Another patient presented with predominantly dystonia and dystonic tremors, associated with spasticity of lower limbs. WES by NGS method, revealed likely pathogenic mutation in gene ZFYVE26, causing SPG 15. Ebrahimi-Fakhari et al[13] described focal dystonia in patients with SPG 15. NGS is helpful in diagnosis in such cases considering the overlap of the neurological symptoms (which is seen in Stevanin G et al. study, Narendiran S. et al. study). [5], [3]
On MRI brain with spine study ([Table 4]), thin corpus callosum is most commonly found in our study (4 cases 23.52%). We found TCC only in SPG11 cases. It is consistent with a comprehensive genetic evaluation of an Italian cohort of patients, da Graça FF et al study. [14] In that study, patients with HSP-TCC (n = 61) were studied and found SPG11 to be the most frequent subtype (26.2%), followed by SPG15 (14.8%), SPG35 (5%), and SPG48 (3%) (13). Other less frequent causes of HSP-TCC are SPG 4, 7, 18, 21, 46, 47, 49, and 54.
The most classic neuroimaging finding “ear-of-the-lynx sign” is characterized by hyperintense on T2-FLAIR-weighted and hypointense on T1-weighted images at the forceps minor of the corpus callosum (genum fibres). This radiological sign may also be present in SPG 15, another form of complicated AR HSP. We have seen Ear of lynx sign in 2 (11.7%) patients, who are SPG 11, consistent with the other studies by da Graça FF et al.[14] and Stevanin G et al.[5] Dorsal spinal cord atrophy, next most common finding in MRI in our cohort, found in 3 (17.64%) patients, (SPG 7, SPG 10 and SPG 30). Dorsal spinal cord atrophy is characteristic but not exclusive for HSP.
In our study, we identified SPG gene loci mutation to be vous in 10, Likely pathogenic in 9, pathogenic in 2 patients. Based on WES by NGS method, patients with hsp phenotype were diagnosed as pathogenic in 40 % (10/25), with vous in 28% (7/25). WES is negative in 8 (31%) cases. In a polish study by Elert- Dobkowska E et al.,[2] 18 pathogenic and likely pathogenic variants in spastic paraplegia probands, as well as six variants of uncertain significance were found. Cao, Y et al.[10] studied a total of 270 patients with HSP phenotype. According to this study, 60 percent diagnoses could be made out of 270 patients, of whom 132(48 percent) cases were diagnosed on NGS and remaining 30 cases were diagnosed with MLPA analysis. In our study, VOUS and WES negative cases could not be further confirmed by MLPA analysis, familial coseggregation or RNA sequencing due to financial constraints. It is a fallacy of our study.
In our cohort, total of 15 novel mutations were seen. In one study by Zeyu Zhu et al.,[15] total of 34 different mutations in the SPAST gene were identified, of which 10 were novel. Our studied identified 15 novel mutations. Hereditary spastic paraplegia is a hereditary neurodegenerative disease, with a rapidly expanding list of genes causing it. Gene causing HSP has been named as SPG with number next to it, in the order of discovery. In our study, most common inherited HSP was found to be SPG 11 which has autosomal recessive inheritance. This similar pattern of was also observed in other studies of Elert-Dobkowska E et al., [2] Stevanin G et al., [5] Narendiran S. et al.,[3] Kara et al.,[6] where SPG11 was found to be commonest ([Table 5]). Autosomal dominant inheritance found to be common in Ishiura H et al study.[15] Luo Y et al.,[7] studied autosomal dominant HSP and SPG 4 was seen as most common cause in china. Worldwide Autosomal dominant inheritance is studied to be the most common pattern of inheritance in HSP. In our study, the reason for more common autosomal recessive inheritance may be the cultural practice of consangunious marriage in Indian ethnicity.
Two patients from different unrelated families (one was from Maharashtra, other from Uttar Pradesh) were detected with Mutation in SPG 11 gene at c.6100C>T p.Arg2034* in our study. In a study by Stevanin G et al.,[5] same variant with mutation at c.6100C>T p.Arg2034* was found in 4 different north African families. This could be indicative of founder effect for this mutation. However, Haplotype analysis may be required for further confirmation.
One more similar mutation was found in 2 unrelated families. A novel missense autosomal recessive mutation in SPG11 gene c.3623C>T (p.Pro1208Leu) loci was found in 2 different patients, one from Maharashtra, other from Uttar Pradesh).
Of the 17 cases in our study, 7 were SPG 11 cases. On further analysis of these cases, age of onset was 1st decade in 3 patients, 2nd decade in 4 patients, ranging from 5 years to 17 years. In Stevanin et al. study, [5] of the 38 patients from 20 families, age of onset was in the range from 2 to 27. We observed male predominance (M: F - 4: 3). In the study by Stevanin et al.,[5] SPG 11 was seen in 18 Males and 20 female patients .Consanguinity is seen in 3 families and family history is found in 1 patients among these families.. Clinical features associated with lower limb weakness and gait disturbances were studied, more commonly observed were cognitive decline in 4, dystonia in 2, seizure in 1, dysarthria in 1 case. Comparing with Stevanin et al. study, [5] of the 38 patients, most common associated neurological feature was cognitive decline in concordance with our study. Dysarthria was frequently seen in their study (16/38). NCS was normal in all cases in our study, but in Stevanin et al. study, NCS showed neuropathy in 13 of 16 cases on follow up. MRI brain showed thin corpus callosum in 3 cases, ear of lynx in 2 patients, bilateral ventricular hyperintensities in 1 and normal in 1 case. More common thin corpus callosum and presence of characteristic ear of lynx sign in SPG 11, is in concordance with the Stevanin et al. study. So, in an HSP suspected case, with onset before 2nd decade, with cognitive decline and thin corpus callosum on mri brain, one should always suspect SPG11. [5]
Conclusion
In conclusion, there is a consider overlap of SPG genes with other neurological diseases like HSMN, HMN2C, FAMILIAL ADULT MYOCLONUS 7. SPG 28 can present with spastic paraparesis and distal upper limb amyotrophy,i.e., SILVER SYNDROME. Next generation sequencing method allowed us for confirming the diagnosis of HSP in patients with multiaxial neurological involvement. One should always suspect, SPG 11in an HSP suspected case with onset before the second decade, cognitive impairment, and a thin corpus callosum on MRI brain.
This study of HSP highlights that genetic diagnosis can be reached with genetic testing by NGS method in patients with multiaxial neurological involvement and confirms the more common prevalence of complicated AR HSP in Indian population, predominantly at SPG 11 loci.
Limitations of study
The main limitation of the study is that parental seggregation is not done ,we have studied only proband and not studed parents.
Source of Funding
None.
Conflict of Interest
None.
References
- Awuah W, Tan J, Shkodina A, Ferreira T, Adebusoye F, Mazzoleni A. Hereditary Spastic Paraplegia: Novel Insights into the Pathogenesis and Management. SAGE Open Med. 2023;12. [Google Scholar] [Crossref]
- Elert-Dobkowska E, Stepniak I, Krysa W, Ziora-Jakutowicz K, Rakowicz M, Sobanska A. Next-Generation Sequencing Study Reveals the Broader Variant Spectrum of Hereditary Spastic Paraplegia and Related Phenotypes. Neurogenetics. 2019;20(1):27-38. [Google Scholar]
- Ishiura H, Takahashi Y, Hayashi T, Saito K, Furuya H, MW. Molecular epidemiology and clinical spectrum of hereditary spastic paraplegia in the Japanese population based on comprehensive mutational analyses. J Hum Genet. 2014;59(3):163-72. [Google Scholar]
- Luo Y, Chen C, Zhan Z, Wang Y, Du J, Hu Z. Mutation and clinical characteristics of autosomal-dominant hereditary spastic paraplegias in China. Neurodegener Dis. 2014;14(4):176-83. [Google Scholar]
- Narendiran S, Debnath M, Shivaram S, Kannan R, Sharma S, Christopher R. Novel insights into the genetic profile of hereditary spastic paraplegia in India. J Neurogenet. 2022;36(1):21-31. [Google Scholar]
- Stevanin G, Azzedine H, Denora P, Boukhris A, Tazir M, Lossos A. Mutations in SPG11 Are Frequent in Autosomal Recessive Spastic Paraplegia with Thin Corpus Callosum, Cognitive Decline and Lower Motor Neuron Degeneration. Brain. 2007;131(3):772-84. [Google Scholar]
- Kamate M, Detroja M. Clinico-Investigative Profile of Hereditary Spastic Paraplegia in Children. Ann Indian Acad Neurol. 2019;22(3):341-4. [Google Scholar]
- Kara E, Tucci A, Manzoni C, Lynch DS, Elpidorou M, Bettencourt C. Genetic and phenotypic characterization of complex hereditary spastic paraplegia. Brain. 2016;139(7):1904-18. [Google Scholar]
- Meyyazhagan A, Bhotla HK, Pappuswamy M, Orlacchio A. The Puzzle of Hereditary Spastic Paraplegia: From Epidemiology to Treatment. Int J Mol Sci. 2022;23(14). [Google Scholar] [Crossref]
- Cao Y, Zheng H, Zhu Z, Yao L, Tian W, Cao L. Clinical and Genetic Spectrum in a Large Cohort of Hereditary Spastic Paraplegia. Mov Disord. 2024;39(4):651-62. [Google Scholar]
- Shi Y, Wang A, Chen B, Wang X, Niu S, Li W. Clinical Features and Genetic Spectrum of Patients with Clinically Suspected Hereditary Progressive Spastic Paraplegia. Front Neurol. 2022;13. [Google Scholar] [Crossref]
- Tesson C, Nawara M, Salih M, Rossignol R, Zaki M, Balwi M. Alteration of Fatty-Acid-Metabolizing Enzymes Affects Mitochondrial Form and Function in Hereditary Spastic Paraplegia. Am J Hum Genet. 2012;91(6):1051-64. [Google Scholar]
- Ebrahimi-Fakhari D, Alecu J, Blackstone C, MA, JF, Mirzaa G. . Spastic Paraplegia 15. Synonyms: Hereditary Spastic Paraplegia Type 15, HSP-ZFYVE26, SPG15, ZFYVE26-Related Hereditary Spastic Paraplegia. 2021. [Google Scholar]
- Graça FD, Rezende TD, Vasconcellos L, Pedroso J, Barsottini O, França M. Neuroimaging in Hereditary Spastic Paraplegias: Current Use and Future Perspectives. Front Neurol. 2019;9. [Google Scholar] [Crossref]
- Zhu Z, Zhang C, Zhao G, Liu Q, Zhong P, Zhang M. Novel Mutations in the SPAST Gene Cause Hereditary Spastic Paraplegia. Parkinsonism Relat Disord. 2019;69:125-33. [Google Scholar] [Crossref]
Article Metrics
- Visibility 10 Views
- Downloads 4 Views
- DOI 10.18231/pjms.v.15.i.1.248-255
-
CrossMark

- Citation
- Received Date May 23, 2024
- Accepted Date September 24, 2024
- Publication Date March 13, 2025